Concepto farmacología
La farmacología es la ciencia que estudia el origen, las acciones y las propiedades que las sustancias químicas ejercen sobre los organismos vivos. En un sentido más estricto se considera la farmacología como el estudio de los fármacos, sea que ésas tengan efectos beneficiosos o bien tóxicos. La farmacología tiene aplicaciones clínicas cuando las sustancias son utilizadas en el diagnóstico, prevención, tratamiento y alivio de síntomas de una enfermedad.
También se puede hablar de Farmacología como el estudio unificado de las propiedades de las sustancias químicas y de los organismos vivientes y de todos los aspectos de sus interacciones, orientado hacia el tratamiento, diagnóstico y prevención de las enfermedades.
La farmacología es una de las ciencias farmacéuticas principales, siendo una aplicación química de una mezcla entre biología molecular, fisiología/fisiopatología, biología celular y bioquímica. Aunque la farmacología destaque sobre las otras ciencias farmacéuticas, no es más importante que las otras (química farmacéutica, farmacognosia, botánica farmacéutica, fitoquímica...). Todas ellas se apoyan mutúamente y resulta muy infrecuente que una trabaje aislada de las demás.
Destino de los fármacos en el organismo
Cualquier sustancia que interactúa con un organismo viviente puede ser absorbida por éste, distribuida por los distintos órganos, sistemas o espacios corporales, modificada por procesos químicos y finalmente expulsada.La farmacología estudia estos procesos en la interacción de fármacos con el hombre y animales, los cuales se denominan:
- Absorción
- Distribución
- Metabolismo
- Excreción
El organismo tiene tejidos susceptibles de ser afectados por un fármaco. Este sistema, órgano o tejido se denomina susceptible o "blanco" de dicho fármaco.
Para que el fármaco ejerza su acción sobre este blanco, debe, generalmente, ser transportado a través de la circulación sanguínea.
Absorción
Para llegar a la circulación sanguínea el fármaco debe traspasar alguna barrera dada por la vía de administración, que puede ser: cutánea, subcutánea, respiratoria, oral, rectal, muscular, via otica, via oftalmica, via sublingual. O puede ser inoculada directamente a la circulación por la vía intravenosa. La farmacología estudia la concentración plasmática de un fármaco en relación con el tiempo transcurrido para cada vía de administración y para cada concentración posible, así como las distintas formas de uso de estas vías de administración.Distribución
Una vez en la corriente sanguínea, el fármaco, por sus características de tamaño y peso molecular, carga eléctrica, pH, solubilidad, capacidad de unión a proteínas se distribuye entre los distintos compartimientos corporales. La farmacología estudia como estas características influyen en el aumento y disminución de concentración del fármaco con el paso del tiempo en distintos sistemas, órganos, tejidos y compartimientos corporales, como por ejemplo, en el líquido cefalorraquideo, o en la placenta.Metabolismo o biotransformación
Muchos fármacos son transformados en el organismo debido a la acción de enzimas.Esta transformación puede consistir en la degradación; (oxidación, reducción o hidrólisis), donde el fármaco pierde parte de su estructura, o en la síntesis de nuevas sustancias con el fármaco como parte de la nueva molécula (conjugación). El resultado de la biotransformación puede ser la inactivación completa o parcial de los efectos del fármaco, el aumento o activación de los efectos, o el cambio por nuevos efectos dependientes de las características de la sustancia sintetizada. La farmacología estudia los mecanismos mediante los cuales se producen estas transformaciones, los tejidos en que ocurre, la velocidad de estos procesos y los efectos de las propias drogas y sus metabolitos sobre los mismos procesos enzimáticos.
Excreción
Finalmente, el fármaco es eliminado del organismo por medio de algún órgano excretor. Principalmente está el hígado y el riñón, pero también son importantes la piel, las glándulas salivales y lagrimales. Cuando un fármaco es suficientemente hidrosoluble, es derivado hacia la circulación sanguínea, por la cual llega a los riñones y es eliminado por los mismos procesos de la formación de la orina: filtración glomerular, secreción tubular y reabsorción tubular. Si el fármaco, por el contrario, es liposoluble o de tamaño demasiado grande para atravesar los capilares renales, es excretada en la bilis, llegando al intestino grueso donde puede sufrir de la recirculación enterohepática, o bien ser eliminado en las heces. La farmacología estudia la forma y velocidad de depuración de los fármacos y sus metabolitos por los distintos órganos excretores, en relación con las concentraciones plasmáticas del fármaco.El efecto de los fármacos, después de su administración, depende de la variabilidad en la absorción, distribución, metabolismo y excreción.
Para que el fármaco alcance su sitio de acción depende de varios factores.
- 1) La tasa y grado de absorción a partir del sitio de aplicacion.
- 2) La tasa y grado de distribución en los líquidos y tejidos corporales.
- 3) La tasa de biotransformación a metabolitos activos o inactivos
- 4) La tasa de excreción.
Acción de los fármacos sobre el organismo
Al estudio del conjunto de efectos sensibles y/o medibles que produce un fármaco en el organismo el hombre o los animales, su duración y el curso temporal de ellos, se denomina farmacodinámica.Para este estudio, la farmacología entiende al sistema, órgano, tejido o célula destinatario del fármaco u objeto de la sustancia en análisis, como poseedor de receptores con los cuales la sustancia interactúa.
La interacción entre sustancia y receptor es un importante campo de estudio, que, entre otros aspectos, analiza:
- Cuantificación de la interacción droga/receptor.
- Regulación de los receptores, ya sea al aumento, disminución o cambio en el nivel de respuesta.
- Relación entre dosis y respuesta.
De acuerdo al tipo de efecto preponderante de un fármaco, farmacodinámicamente se les clasifica en:
- Agonistas farmacológicos, si produce o aumenta el efecto.
- Antagonistas farmacológicos, si disminuye o elimina el efecto.
También existe un campo especial de estudio de los efectos farmacológicos de sustancias durante la gestación.
En el hombre, los efectos sobre el embrión y el feto de los fármacos es un campo de intenso estudio.
Ramas de la farmacología
- Farmacodinamia: ciencia que estudia el mecanismo de acción de los fármacos, es decir estudia como los procesos bioquímicos y fisiológicos dentro del organismo se ven afectados por la presencia del fármaco
- Farmacognosia: estudia todas las drogas desde el punto de vista farmacéutico, es decir como sustancias naturales de actividad biológica, sus fuentes y características.
- Química farmacéutica: estudia los fármacos desde el punto de vista químico, lo que comprende el descubrimiento, el diseño, la identificación y preparación de compuestos biológicamente activos, la interpretación de su modo de interacción a nivel molecular, la construcción de su relación estructura-actividad y el estudio de su metabolismo.
- Farmacotecnia: rama encomendada a la preparación de las drogas según administración individualizada.
- Toxicología: el estudio de los efectos nocivos o tóxicos de los fármacos
- Biofarmacia: el estudio de la biodisponibilidad de los fármacos
- Posología: el estudio de la dosificación de los fármacos
- Farmacocinética: el estudio de los procesos físico-químicos que sufre un fármaco cuando se administra o incorpora a un organismo. Estos procesos serían liberación, absorción, distribución, metabolización y eliminación
- Farmacología clínica: evalúa la eficacia y la seguridad de la terapéutica por fármacos
- Farmacovigilancia: el estudio de las reacciones adversas que provocan los fármacos sobre el organismo
- Cronofarmacología: El estudio de la correcta administracion de medicamentos conforme al ciclo circadiano del ser humano, esto con el fin de maximizar la eficacia y disminuir los efectos colaterales.
- Terapéutica: el estudio de las interacciones beneficiosas de los fármacos con el organismo
- Otras terapéuticas:
- Farmacognosia: estudio de plantas y drogas que de ellas se derivan
- Farmacobotánica: estudio de plantas medicinales
- Fitoterapia: tratamiento de enfermedades con plantas medicinales.
- Homeopatía: administración de sustancias de origen animal, vegetal o mineral; en dosis infinitesimales de dilución, produciría una sintomatología opuesta a la generada por la utilización de la sustancia en estado puro.
Farmacodinamia
En farmacología, la farmacodinámica, farmacosis o farmacodinamia, es el estudio de los efectos bioquímicos y fisiológicos de los fármacos y de sus mecanismos de acción y la relación entre la concentración del fármaco y el efecto de éste sobre un organismo. Dicho de otra manera: el estudio de lo que le sucede al organismo por la acción de un fármaco. Desde este punto de vista es opuesto a lo que implica la farmacocinética: lo que le sucede al fármaco por la acción del organismo.
La farmacodinámica puede ser estudiada a diferentes niveles, es decir, sub-molecular, molecular, celular, a nivel de tejidos y órganos y a nivel del cuerpo entero, usando técnicas
Historia
Desde que se inició el estudio de la acción de los fármacos, se observó que ésta aumentaba de forma proporcional a la dosis del fármaco administrado, hasta llegar a un máximo, punto a partir del cual no aumentaba por más que aumentara la cantidad de fármaco. Esto hizo pensar que los fármacos actuaban sobre unos "sitios" específicos en el organismo. Estos sitios son limitados, lo que explicaba el comportamiento del fármaco: aumenta la acción conforme se van ocupando los sitios, pero cuando están todos ocupados se estabiliza. Esto abrió paso al concepto de sitios receptivos específicos, o receptores. Estos receptores son estructuras celulares que tienen una finalidad concreta y que son activados en su actuación por distintas sustancias, tanto naturales como externas al organismo (fármacos). Por tanto, los fármacos no crean efectos nuevos en el organismo, limitándose a potenciar o inhibir efectos ya existentes.
El conocimiento de estos efectos y de los receptores responsables de los mismos ha sido el eje de la investigación farmacodinámica.
Tipos de efectos farmacológicos
Al administrar una droga se pueden conseguir diversos efectos medicamentosos que se correlacionan con la acción del fármaco.
- Efecto primario: es el efecto fundamental terapéutico deseado de la droga.
- Efecto placebo: son manifestaciones que no tienen relación con alguna acción realmente farmacológica.
- Efecto indeseado: cuando el medicamento produce otros efectos que pueden resultar indeseados con las mismas dosis que se produce el efecto terapéutico;
- Efecto colateral: son efectos indeseados consecuencia directa de la acción principal del medicamento.
- Efecto secundario: son efectos adversos independientes de la acción principal del fármaco.
- Efecto tóxico: por lo general se distingue de los anteriores por ser una acción indeseada generalmente consecuencia de una dosis en exceso. Es entonces dependiente de la dosis, es decir, de la cantidad del medicamento al que se expone el organismo y del tiempo de exposición.
- Efecto letal: acción biológica medicamentosa que induce la muerte.
Dosis
La dosis es la cantidad de una droga que se administra para lograr eficazmente un efecto determinado. El estudiar o estimar la dosis efectiva y la forma correcta de administración del fármaco se le llama dosificación, administrada por la posología. La dosis puede clasificarse en:- Dosis subóptima o ineficáz: es la máxima dosis que no produce efecto farmacológico apreciable.
- Dosis mínima: es una dosis pequeña y el punto en que empieza a producir un efecto farmacológico evidente.
- Dosis máxima: es la mayor cantidad que puede ser tolerada sin provocar efectos tóxicos.
- Dosis terapéutica: es la dosis comprendida entre la dosis mínima y la dosis máxima.
- Dosis tóxica: constituye una concentración que produce efectos indeseados.
- Dosis mortal: dosis que inevitablemente produce la muerte.
- DL50: denominada Dosis Letal 50 o Dosis Mortal 50%, es la dosis que produce la muerte en 50% de la población que recibe la droga. Así también se habla con menos frecuencia de DL20, DL90 y DL99.
- DE50: denominada Dosis Efectiva 50 es la dosis que produce un efecto terapéutico en el 50% de la población que recibe la droga.
Selectividad
El estudio de los mecanismos de acción de un medicamento sobre las células comienza conociendo la selectividad de la droga. Algunos medicamentos tienen una muy baja selectividad por lo que ejercen sus efectos sobre muchos órganos y tejidos, mientras que otras drogas son altamente selectivos, como un antiácido que ejerce su función en células de un órgano específico. Para la mayoría de las drogas, la acción que ejercen sobre el cuerpo es críticamente dependiente de su estructura química, de tal modo que variaciones minúsculas en esa estructura altera tremendamente la selectividad del medicamento. selectividad: "efecto especifico organico" naturaleza. dosis, intervalo, tipo de paciente/caracteristicas individuales.Receptores
La mayoría de las drogas ejercen su acción sobre una célula por virtud de su reconocimiento de receptores sobre la superficie celular, específicamente por tener la configuración molecular que se ajusta al dominio de unión del receptor. La selectividad de un fármaco por uno o varios órganos se fundamenta principalmente por lo específico que es la adherencia del medicamento al receptor diana. Algunos fármacos se unen a un solo tipo de receptores, mientras que otros tienen la facultad bioquímica de unirse a mutliples tipos de receptores celulares..
Agonistas
Agonista: Es una droga que produce un efecto combinándose y estimulando al receptor, estos pueden ser clasificados como:-Agonistas Completos: los que producen la máxima respuesta posible.
-Agonistas Parciales: son los agonistas que no logran alcanzar el Emax de los agonistas completos.
-Agonistas Inversos: los que logran efectos opuestos a los producidos por los agonistas completos y parciales.
Antagonistas
Antagonista: Es una droga que produce efecto farmacológico bloqueando al receptor y por lo tanto es capaz de reducir o abolir el efecto de los agonistas. Los antagonistas pueden ser clasificados como:1. Antagonistas competitivos: Son aquellos que bloquean el efecto de los agonistas compitiendo por el mismo sitio de fijación en el receptor. Hay dos tipos básicos:
1.1.- Antagonistas reversibles: que pueden ser desplazados del receptor por dosis crecientes del agonista (antagonismo superable). Los antagonistas competitivos reversibles desplazan la curva dosis-respuesta de los agonistas hacia la derecha (es decir aumentan la DE50 y reducen la afinidad) sin afectar la Emax y eficacia del agonista.
1.2. Antagonistas irreversibles: que no pueden ser desplazados del receptor por dosis crecientes del agonista (antagonismo insuperable). Los antagonistas competitivos irreversibles reducen la Emax y la eficacia del agonista.
2. Antagonistas no competitivos: Son aquellos que bloquean el efecto de los agonistas uniéndose al receptor en un sitio distinto al sitio de fijación del agonista. Estos antagonistas reducen el Emax ( la eficacia ). Pueden ser a su vez:
2.1 Reversibles: Los cuales se disocian fácilmente del receptor al suspender su administración en el paciente o el lavado del tejido aislado.
2.2.- Irreversibles: que se fijan permanentemente o modifican covalente el receptor el cual queda permanentemente inutilizado y tiene que ser reemplazado por uno nuevo.
Afinidad y actividad intrínseca
En la interacción del fármaco—y, en realidad, cualquier ligando—tiene dos propiedades, la afinidad, que es la capacidad del medicamento de establecer una unión estable, y la actividad intrínseca, que es la eficacia biológica del complejo droga:receptor en producir una mayor o menor respuesta celular. De modo que algunos medicamentos pueden tener la misma afinidad estructural por un receptor, más uno puede tener una gran eficacia en la unión, mientras que el otro mucho menor. Un agonista y un antagonista pueden tener la misma afinidad por el receptor, pero el antagonista no tiene eficacia en producir actividad intrínseca en la célula como consecuencia de su unión con el receptor.Efectos en el cuerpo
La mayoría de los fármacos actúan inhibiendo o estimulando las células, destruyéndolas o reemplazando en ellas determinadas sustancias. Los mecanismos de acción se fundamentan principalmente en su asociación con receptores asociados a canales iónicos, a una proteína G, receptores con actividad enzimática intrínseca o con receptores asociados a proteínas enzimáticas como la tirosincinasa.Control de canales iónicos
El contacto con receptores asociados a canales iónicos aumenta la permeabilidad de la membrana y la conducción de iones a través de la membrana plasmática alterando su potencial de membrana eléctrico facilitando su despolarización.Formación de segundos mensajeros
La formación de segundos mensajeros acoplados a una proteína G activa enzimas como la adenilciclasa, el AMP cíclico, proteíncinasas, las cuales transducen señales que inducen gran cantidad de posibles efectos funcionales sobre la célula. Otras moléculas diana de un gran número de fármacos son las pertenecientes al sistema de los fosfoinosítidos de la membrana celular. Ellos también son acoplados a segundos mensajeros y ejercen respuestas celulares por medio del calcio, por ejemplo.Actividad enzimática intrínseca
Cuando una droga se une a su receptor tiende a ejercer control directo sobre la fosforilación de proteínas celulares, modificando la estructura conformacional de la proteína, activando o inactivandola.Control de transcripción
Algunos medicamentos atraviesan la membrana plasmática y actúan directamente sobre el núcleo celuar y sobre receptores intracelulares, revirtiendo la represión del ADN y aumentando la transcripción y síntesis protéica.Farmacodinámica multicelular
El concepto de la farmacodinámica ha sido ampliado para poder incluir a la Farmacodinámica Multicelular (MCPD), que estudia las propiedades estáticas, dinámicas y las relaciones entre un conjunto de fármacos y una organización multicelular dinámica y diversa cuatridimensional. Es el estudio de todo el funcionamiento del fármaco en el mínimo sistema multicelular (mMCS).Nuevos fármacos
Los estudios farmacodinámicos pueden ayudar al desarrollo racional de agentes farmacéuticos mediante los siguientes aspectos:- Demostrar la actividad biológica del fármaco sobre su diana terapéutica cuando se administra a los pacientes.
- Analizar los efectos moleculares y biológicos que se producen como consecuencia de la acción del fármaco sobre la diana
- Mediante estos estudios se puede analizar un rango de Dosis Biológica Óptima del fármaco, es decir, la dosis mínima de fármaco que produce el máximo efecto biológico, y explorar la eficacia de los distintos esquemas de administración en función del efecto biológico que produce cada uno de ellos.
- Identificar los efectos moleculares (marcadores) relacionados con la respuesta y resistencia al fármaco.
Farmacocinética
La farmacocinética es la rama de la farmacología que estudia los procesos a los que un fármaco es sometido a través de su paso por el organismo. Trata de dilucidar qué sucede con un fármaco desde el momento en el que es administrado hasta su total eliminación del cuerpo.Para ello, se han desarrollado diferentes modelos que simplifiquen los numerosos procesos que tienen lugar entre el organismo y el fármaco. Aún cuando dentro de los mismos el modelo policompartimental es el más próximo a la realidad, la complicación que conlleva ha hecho que sean los modelos monocompartimental y en todo caso el bicompartimental los más usados. Desde esos prismas, el estudio detallado de los sucesivos pasos que atraviesa el fármaco en el organismo, se agrupan bajo el anagrama LADME:
- Liberación del producto activo,
- Absorción del mismo,
- Distribución por el organismo,
- Metabolismo o inactivación, al ser reconocido por el organismo como una sustancia extraña al mismo, y
- Eliminación del fármaco o los residuos que queden del mismo.
Todos estos conceptos se pueden representar mediante fórmulas matemáticas que tienen su correspondiente representación gráfica. De esta manera se puede conocer tanto las características de una molécula, así como la manera en que se comportará determinado fármaco conociendo algunas de sus características básicas. Así, el conocimiento del pKa, su biodisponibilidad o hidrosolubilidad, orienta sobre su capacidad de absorción o distribución en el organismo.
Las gráficas resultantes del estudio de un fármaco tienen valor trascendente en aplicaciones en la industria (cálculos de bioequivalencia en el diseño de fármacos genéricos, por ejemplo) o en la aplicación clínica de los conceptos farmacocinéticos. En efecto, la farmacocinética clínica provee abundantes pautas de actuación para el correcto manejo de los fármacos, buscando el máximo de efectividad y utilidad para los profesionales de la medicina humana y veterinaria.
Modelos farmacocinéticos
El resultado final de las transformaciones que sufre un fármaco en el organismo y las reglas que las rigen, depende de la suma de múltiples factores habitualmente interrelacionados entre sí. Con el objetivo de simplificar el estudio se diseñaron modelos de funcionamiento basados fundamentalmente en la consideración del organismo como compartimentos relacionados entre sí. Conceptualmente, la propuesta más simple es la consideración homogénea del organismo, con la existencia de un solo compartimento. Este modelo monocompartimental presupone que las concentraciones plasmáticas del fármaco son fiel reflejo de las concentraciones en otros fluidos o tejidos, y que la eliminación del fármaco es directamente proporcional a los niveles en el organismo del fármaco (cinética de primer grado).Sin embargo, no siempre estos presupuestos recogen con una fidelidad aproximada lo que ocurre realmente en el organismo. Por ejemplo, no todos los tejidos presentan la misma riqueza en aporte sanguíneo, por lo que en unos la distribución del fármaco será más lenta que en otros. Además, existen algunos tejidos (como por ejemplo el tejido del cerebro) que presentan una verdadera barrera a la llegada de los fármacos, que será saltada con mayor o menor facilidad dependiendo de las características del fármaco. De modo que, manteniendo los otros condicionantes de proporcionalidad entre los distintos tejidos y de la velocidad de eliminación, el organismo se podría comportar como dos compartimentos: uno al que podemos llamar compartimento central que presenta una velocidad de distribución más elevada y constituido por los órganos y sistemas más intensamente irrigados y un compartimento periférico constituido por los órganos menos irrigados, quedando algunos tejidos como el cerebro en una posición variable según la facilidad que presente el fármaco para atravesar la barrera que lo separa de la sangre.
{kind=link}
Este modelo bicompartimental será diferente considerando en cual compartimento se produce la eliminación. Lo más frecuente es que la misma se produzca en el compartimento central, ya que hígado y riñones son órganos muy bien irrigados. No obstante, puede darse la situación de que la eliminación se realice desde el compartimento periférico o incluso desde ambos. Obtenemos así al menos tres variedades de modelo bicompartimental, que sin embargo sigue sin explicarnos todas las posibilidades.
Modelo monocompartimental
Se le conoce como farmacocinética lineal porque al graficar la relación los distintos factores implicados (dosis, concentraciones en el plasma sanguíneo, eliminación, etcétera) la representación gráfica es una recta o una aproximación a ella. Es muy útil para fármacos que se distribuyen con rapidez desde el plasma a otros fluidos y tejidos.El cambio de concentración respecto al tiempo viene dado por C=Cinicial*E^(-kelt) que si se representa lnC frente tiempo da lugar a una recta.
Modelos policompartimentales
Al graficar la relación entre los distintos factores, la imagen resultante es una curva, siendo entonces necesario el cálculo de determinadas áreas bajo esa curva para hallar los resultados a las interrelaciones presentadas. Por ello, estos modelos reciben también el nombre de farmacocinética no lineal, y se basa de forma muy importante en la cinética de Michaelis-Menten. Los factores de no linealidad de una reacción, serían, entre otros, los siguientes:{kind=link}
- Absorción polifásica: La absorción del fármaco sigue al menos dos picos de máxima intensidad, con lo que mediatiza la linealidad de su llegada al plasma.
- La naturaleza del fármaco hace clara distinción entre tejidos de alta y baja irrigación.
- Saturación enzimática: En fármacos en los que su eliminación es dependiente de su biotransformación, al aumentar la dosis, las enzimas responsables de su metabolismo se saturan y la concentración plasmática del fármaco aumenta desproporcionalmente, por lo que su depuración deja de ser constante.
- Inducción o inhibición enzimática: Algunos fármacos tienen la capacidad de inhibir o estimular su propio metabolismo, en una reacción de retroalimentación. Tal es el caso de fluvoxamina, fluoxetina y fenitoína. Al administrar mayores dosis de estos medicamentos, las concentraciones plasmáticas de fármaco sin metabolizar aumenta y el tiempo medio de eliminación aumenta con el tiempo. Por esa razón, para fármacos con farmacocinética no-lineal, es necesario ajustar la posología o régimen en casos de incrementar la dosis.
- El riñón establece mecanismos activos de eliminación para algunos fármacos, independientes de los niveles de concentración plasmática.
Biodisponibilidad
A efectos prácticos, se define la biodisponibilidad de un fármaco como la fracción del mismo que alcanza la circulación sistémica del paciente. O dicho de otra manera, el porcentaje de fármaco que aparece en plasma. Desde este prisma, la administración de un fármaco por vía intravenosa presentaría la mayor biodisponibilidad posible, por lo que se considera la unidad (o el 100%). A partir de aquí, la biodisponibilidad se calcula comparando la vía a estudiar con respecto a la vía intravenosa («biodisponibilidad absoluta») o a un valor estándar de otras presentaciones del fármaco en estudio («biodisponibilidad relativa»).![B_A = \frac{[ABC]_P . D_{IV}}{[ABC]_{IV} . D_P}](http://upload.wikimedia.org/math/5/c/a/5ca3c5293ac76c5cc118262203196262.png)
![\mathit B_R = \frac{[ABC]_{A} . dosis_{B}}{[ABC]_{B} . dosis_{A}}](http://upload.wikimedia.org/math/8/9/1/89142147383556850b8ddeefa9d174b1.png)
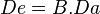 (en donde De es la dosis eficaz, B la biodisponibilidad y Da la dosis administrada) podemos calcular la cantidad de fármaco en plasma que realmente tiene capacidad para realizar su efecto.
(en donde De es la dosis eficaz, B la biodisponibilidad y Da la dosis administrada) podemos calcular la cantidad de fármaco en plasma que realmente tiene capacidad para realizar su efecto.Así, si tenemos un fármaco cuya biodisponibilidad es de 0,8 (o del 80%) y se administra una dosis de 100 mg, la ecuación se resolvería:
Diferentes formas de comprimidos, los cuales conllevan diferentes comportamientos farmacocinéticos después de su administración.
- Forma galénica
- Forma química
- Vía de administración
- Estabilidad
- Metabolización

donde Q sería la constante de pureza del fármaco.

Finalmente, por la ecuación de Henderson-Hasselbalch, y sabiendo el
 del fármaco (pH al cual presenta equilibrio entre sus moléculas ionizadas y no ionizadas), podemos calcular la cantidad de fármaco no ionizado, y, por tanto, la cantidad de fármaco objeto de la absorción:
del fármaco (pH al cual presenta equilibrio entre sus moléculas ionizadas y no ionizadas), podemos calcular la cantidad de fármaco no ionizado, y, por tanto, la cantidad de fármaco objeto de la absorción:
El acrónimo LADME
Una vez que el fármaco entra en contacto con el organismo, suceden varias fases que se reconocen con el acrónimo LADME:- Liberación de la sustancia activa,
- Absorción de la misma por parte del organismo
- Distribución por el plasma y los diferentes tejidos,
- Metabolización, es decir inactivación de una sustancia xenobiótica y, finalmente,
- Excreción o eliminación de la sustancia o de los productos de su metabolismo.
Cada una de las fases está sujeta a las interacciones físico-químicas entre fármaco y organismo, que se pueden expresar de forma matemática. La farmacocinética, pues, se apoya en ecuaciones matemáticas que permiten predecir el comportamiento del fármaco, y que dan cuenta, de una forma preferente, de la relación que existe entre las concentraciones plasmáticas y el tiempo transcurrido desde la administración.
Liberación
La liberación es el primer paso del proceso en el que el medicamento entra en el cuerpo y libera el contenido del principio activo administrado. El fármaco debe separarse del vehículo o del excipiente con el que ha sido fabricado, y para algunos autores comprende tres pasos: desintegración, disgregación y disolución. Se hace una especial referencia a la ionización de las moléculas del fármaco como factor limitante de la absorción, debido a las propiedades de las membranas celulares que dificultan el paso a su través de moléculas ionizadas. La recomendación de masticar los comprimidos o tabletas realizada por muchos profesionales radica, precisamente, en facilitar esta fase, en concreto la disgregación.En todo caso, es necesario recordar que las características de los excipientes tienen un papel fundamental, ya que tienen como una de sus funciones el crear el ambiente adecuado para que el fármaco se absorba correctamente. Es por ello que medicamentos con la misma dosis, pero de distintas marcas comerciales pueden tener distinta bioequivalencia, es decir, alcanzan concentraciones plasmáticas distintas, y, por tanto, efectos terapéuticos diferentes.
Disolución
En una situación típica, al ingerir una tableta pasa por el esófago al estómago. Por razón de que el estómago tiene un ambiente acuoso, es el primer lugar donde la tableta se disolverá. La velocidad de disolución es un elemento clave en el control de la duración del efecto del fármaco, y por ello, diferentes formas del mismo medicamento pueden tener los mismos ingredientes activos, pero difieren en la velocidad de disolución. Si se administra un fármaco bajo una forma galénica que no es rápidamente disuelta, el fármaco se absorberá más gradualmente en el tiempo, alcanzando una más larga duración en su acción. La consecuencia es una mejora en su complianza, logrando en definitiva, que el medicamento no tenga que ser tomado tan a menudo. Además, una forma de liberación lenta mantendrá concentraciones en rangos terapéuticos aceptables por un período más duradero a diferencia de las presentaciones de liberación rápida, que tienen picos de concentraciones plasmáticas más pronunciados.La velocidad de disolución se describe por la ecuación de Noyes-Whitney:

 es la velocidad de disolución.
es la velocidad de disolución.- A es la área superficial del sólido.
- C es la concentración del sólido en el medio de disolución principal.
- Cs es la concentración del sólido en la capa de difusión que rodea al sólido.
- D es el coeficiente de difusión.
- L es el grosor de la capa de difusión.
Ionización
Las membranas celulares presentan una resistencia al paso de moléculas ionizadas superior a la que presenta a las sustancias no ionizadas y liposolubles. Este hecho es de importancia sobre todo con sustancias que son anfotéricamente débiles. El pH ácido del estómago y la posterior alcalinización del mismo en el intestino, modifican los grados de ionización de ácidos y bases débiles, dependiendo del pKa de cada sustancia. El pKa es el pH en el que una sustancia presenta un equilibrio entre las moléculas ionizadas y las no ionizadas, y para su cálculo es importante considerar la ecuación de Henderson-Hasselbalch.Absorción
La absorción significa atravesar algún tipo de barrera, diferente según la vía de administración usada, pero que en último término se puede reducir al paso de barreras celulares. O dicho de otra forma, la interacción de la molécula con una membrana biológica, donde las características fisicoquímicas, tanto del fármaco como de la membrana, determinarán el resultado del proceso.Membranas biológicas
Esquema de una membrana celular.
Los fosfolípidos son responsables de las características de permeabilidad de la membrana así como eslabón importante en la cadena anabólica de numerosas sustancias de defensa (prostaglandinas, leucotrienos, ...). Suponen aproximadamente un 40% a 45% de los componentes de la membrana.
Por su parte, las proteínas constituyen alrededor del 50% de los constituyentes de las membranas, y le dan la rigidez estructural necesaria a la misma. Además, se comportan como el punto de inicio de las reacciones a las moléculas que llegan hasta la membrana (receptores), las metabolizan (enzimas), transportan moléculas en contra del gradiente de concentración a ambos lados de la membrana (bombas), o crean canales por donde puedan pasar éstas moléculas (proteínas canal).
Finalmente, nos podemos encontrar entre un 7% y un 10% de hidratos de carbono (glucolípidos y glucoproteínas) que actúan como modulador de las proteínas receptores.
El receptor celular es el punto último del viaje del fármaco destinado a lograr un efecto sobre el organismo humano. De las complejas interrelaciones entre ambos se encarga otra disciplina de la farmacología: la farmacodinámica.
Vías de administración
Las barreras que ha de atravesar y las características de la absorción de cada sustancia vienen determinadas por cual haya sido la vía por la que ha llegado la misma a entrar en contacto con el organismo, o dicho de otro modo, de cual sea la vía de administración. Aquí se verá sólo una breve tabla de las diferentes vías de administración, con las características especiales en cada caso de cara a la absorción.La vía oral es la vía recomendada para humanos. Desafortunadamente, no todos los productos pueden adaptarse para su uso por esta vía. En la vía oral el fármaco llega al organismo habitualmente después de la deglución. Una vez en el estómago, se somete a las características de los jugos del mismo, que por su acidez favorece mucho la ionización del fármaco, lo que hace que la absorción sea difícil. A pesar de todo, no son escasos los fármacos que se absorben a nivel de la mucosa gástrica: los muy liposolubles, como el alcohol o ácidos débiles como los salicilatos o los barbitúricos que presentan menores niveles de ionización a pH bajo. Cuando llega el fármaco al intestino delgado cambia el pH luminal y se favorece bastante la absorción pasiva. De hecho, prácticamente todos los fármacos, menos los ácidos y bases fuertes, se absorben a este nivel. Además, en la mucosa intestinal hay numerosos mecanismos para realizar procesos de absorción en contra de gradiente, aunque difícilmente se logran niveles plasmáticos suficientes para que sean efectivos. Esta falta de absorción para algunos fármacos se aprovecha para utilizarlos a nivel local (como la neomicina o los laxantes). Igualmente, por su similitud estructural, se utiliza este efecto para administrar fármacos que no atraviesen la piel y que actúen a nivel local, constituyendo lo que se conoce como vía dérmica o vía tópica.
La vía parenteral ofrece indudables ventajas sobre la vía oral: permite su uso en pacientes que no pueden o no deben deglutir, permite el uso de sustancias polipeptídicas y otras que se inactivan por los jugos gastrointestinales y evitan el primer paso hepático. Sin embargo precisa de instrumental para su realización y presenta inconvenientes como la infección local, tromboflebitis, neuralgias, necrosis dérmicas, etc. Desde el punto de vista farmacodinámico, la principal ventaja es la facilidad para ajustar la dosis eficaz, ya que la biodisponibilidad se considera del 100% en la mayoría de los casos.
Respecto a la vía respiratoria su interés fundamental es que brinda la posibilidad de la utilización de sustancias en estado gaseoso (casi exclusivamente oxígeno o anestésicos generales). La absorción sigue las leyes del intercambio de gases a nivel alveolar y tiene la ventaja de poner en disposición una gran superficie de absorción.
Características de la absorción
Hay que tener presente la existencia de una serie de factores que modifican la absorción:- Solubilidad: la absorción del fármaco es más rápida cuando está en solución acuosa con respecto a si está en solución oleosa, y, a su vez, ambas son más rápidas que la que presentaría en forma sólida.
- Cinética de disolución de la forma farmacéutica del medicamento. De la misma depende la velocidad y la magnitud de la absorción del principio activo.
- Concentración del fármaco: a mayor concentración, mayor absorción.
- Circulación en el sitio de absorción: a mayor circulación, mayor absorción.
- Superficie de absorción: a mayor superficie, mayor absorción.
Absorción pasiva o difusión pasiva
El paso de la sustancia implicada se produce sin gasto de energía, a favor de gradientes de concentración. Puede producirse a través de la membrana propiamente dicha o a través de ciertas proteínas que forman poros.
{kind=link}
- Difusión simple: depende del tamaño de las moléculas, y puede realizarse a través de la bicapa lipídica de la membrana o a través de los poros acuosos constituidos por las proteínas insertas en la misma. Las sustancias no ionizadas tienen mayor facilidad para la misma, siguiendo la ley de Fick, por la cual
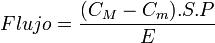
- Difusión facilitada: se debe a la presencia de un gradiente a ambos lados de la membrana para otras moléculas que tienen la propiedad de unirse al fármaco y arrastrarlo en su migración. Son las moléculas facilitadoras, y se incluyen dentro de la difusión pasiva debido a que no consumen energía en su trasiego. Sin embargo, a diferencia de la difusión simple, este mecanismo es saturable, al depender del número de moléculas facilitadoras.
Absorción activa o transporte activo
El paso de la sustancia implica un gasto energético en forma de moléculas de ATP. Permite la absorción contra gradiente y depende también de las moléculas facilitadoras, que en esta ocasión no migran en función de un gradiente, sino gracias al gasto energético. Por tanto es un mecanismo también saturable. Se realiza mediante las proteínas bomba de la membrana (ATP Binding Cassete), teniendo especial transcendencia la MDR1(del inglés MultiDrug Resistence tipo 1) que exporta un gran número de fármacos y es factor clave de la resistencia de las células cancerosas a los quimioterápicos.La endocitosis es un mecanismo propio de algunas células por el que mediante la formación de vesículas originadas a partir de la membrana citoplásmica, introducen en su interior sustancias externas a ellas. Es un mecanismo que consume gran cantidad de energía, pero tiene la ventaja de introducir grandes cantidades de material al interior celular.
Distribución
La distribución de los fármacos puede definirse, entre otras formas, como la llegada y disposición de un fármaco en los diferentes tejidos del organismo. Es un proceso muy importante, toda vez que, según su naturaleza, cada tejido puede recibir cantidades diferentes del fármaco, el cual, además, pasará allí tiempos variables.A la hora de hablar de la distribución, habrá que tener en cuenta los conceptos sobre compartimentación del organismo vistos en el apartado de Modelos farmacocinéticos.
Factores que afectan la distribución
Son múltiples, pero siguiendo a Pascuzzo, los más importantes son los tres siguientes: los volúmenes físicos del organismo, la tasa de extracción y la unión a proteínas plasmáticas y, o, tisulares.Volúmenes físicos del organismo
Este concepto está relacionado con la multicompartimentalización. Considerando los fármacos como solutos, los distintos tejidos con especificidad del organismo van a actuar como los solventes que darán pie a las diferentes concentraciones del fármaco. Así, dependiendo de la naturaleza química de éste, habrá una especial predisposición de las sustancias liposolubles por la grasa corporal o de las hidrosolubles por el líquido extracelular. Este volumen de distribución (Vd) de un fármaco en el organismo es tan sólo aparente, pues conceptualmente se trataría del volumen necesario para contener de forma homogénea en todo el organismo una cantidad determinada de fármaco, que viene dada por el nivel de la concentración del mismo en el plasma. Desde el punto de vista físico el Vd viene determinado por la siguiente fórmula: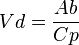 en donde Ab es la cantidad total de fármaco en el cuerpo y Cp la concentración plasmática del mismo.
en donde Ab es la cantidad total de fármaco en el cuerpo y Cp la concentración plasmática del mismo.Siendo la Ab conocida, pues es equivalente a la dosis de fármaco administrada, la fórmula nos indica que la relación existente entre Vd y la Cp es una relación de proporcionalidad inversa. Es decir, que a mayor Cp menor Vd y viceversa. O lo que es lo mismo, que los factores que aumenten la Cp disminuirán el valor del Vd. Esto nos pone sobre la pista de la importancia del conocimiento de las concentraciones plasmáticas del fármaco y de los factores que lo modifican.
Aplicando a esta fórmula los conceptos aprendidos en el apartado de la biodisponibilidad, podemos calcular la cantidad de fármaco a administrar para conseguir una determinada concentración de fármaco en el organismo (dosis de carga):

Este concepto tiene interés clínico, pues a veces necesitamos alcanzar una determinada concentración de fármaco que sabemos es la óptima para que realice sus efectos en el organismo (caso de la digitalización de un paciente).
Tasa de extracción
Se refiere a la proporción del fármaco que es retirado de la circulación por cada órgano, una vez que el flujo sanguíneo lo haya hecho pasar a través de dicho órgano.Este nuevo concepto integra otros anteriores, ya que la tasa de extracción va a depender de distintos factores:- Características del fármaco, entre ellas su pKa.
- Redistribución tisular: En algunos fármacos se produce una distribución rápida e intensa en determinados tejidos, hasta llegar al equilibrio con la concentración plasmática. Sin embargo otros tejidos más lentos continúan retirando fármaco del plasma, con lo que la concentración en el primer tejido queda por encima de la plasmática y por tanto sale fármaco del tejido hacia el plasma. Este fenómeno se sigue sucediendo durante un tiempo hasta alcanzar el equilibrio definitivo. Se obtiene por tanto dos concentraciones del fármaco en el tejido más sensible: una inicial más elevada y otra posterior consecuencia de la redistribución tisular.
- Diferencial de concentración con los tejidos.
- Superficie de intercambio.
- Presencia de barreras naturales. Son obstáculos a la difusión similares a las encontradas en la absorción. Las más interesantes son:
- Permeabilidad de los lechos capilares, que no es igual en todos los tejidos.
- Barrera hematoencefálica: está localizada entre el plasma sanguíneo de los vasos cerebrales y el espacio extracelular del encéfalo. Dificulta la llegada de fármacos al mismo.
- Barrera placentaria: en la mujer embarazada, evita la llegada de gran cantidad de fármacos al feto, que pudieran ser tóxicos para el mismo.
Unión a proteínas plasmáticas
Por estudios realizados in vitro en condiciones ideales, el equilibrio entre la concentración plasmática y tisular del fármaco sólo se ve alterado de forma significativa con índices de fijación a proteínas plasmáticas superiores al 90%. A partir de estos niveles se produce un "secuestro" del fármaco que disminuye su presencia en los tejidos por debajo del 50% del total. Esto es importante a la hora de considerar las interacciones farmacológicas: un fármaco con un índice de fijación a proteínas plasmáticas inferior al 90%, si es desplazado de su unión a las proteínas por otro fármaco no va a aumentar significativamente su presencia en los tejidos. Por el contrario, con índices de unión a proteínas plasmáticas superiores al 95%, pequeños desplazamientos pueden originar importantes modificaciones de la concentración tisular y, por tanto, mayor riesgo de toxicidad por exceso de su efecto en los tejidos.De las proteínas plasmáticas quizás las de más interés sean las albúminas, por su cantidad y su capacidad para unirse a otras sustancias. Otras proteínas de interés son las glicoproteínas, las lipoproteínas y en menor medida las globulinas.
Como podrá comprenderse, situaciones clínicas que supongan modificación de los niveles de proteínas plasmáticas (por ejemplo hipoalbuminemias secundarias a procesos renales) pueden tener transcendencia en el efecto y toxicidad de un fármaco que presente índices de unión a proteínas plasmáticas superiores al 90% (ó 0,9).
Metabolismo o biotransformación
Muchos fármacos son transformados en el organismo debido a la acción de enzimas. Esta transformación, destinada a contrarrestar el posible efecto perjudicial de una sustancia extraña al organismo, es el concepto básico del metabolismo xenobiótico, siendo los fármacos las sustancias xenobióticas por excelencia.Diagrama del metabolismo hepático de fase I y II.
En el humano y en la mayoría de los mamíferos, el metabolismo de los fármacos se realiza fundamentalmente a nivel del hígado. Como resultado de la biotransformación se obtienen nuevas sustancias que reciben el nombre de metabolitos. Los metabolitos pueden mantener la capacidad del fármaco original para ejercer sus efectos, o bien haberla vista disminuida, aumentada o incluso haber cambiado sus efectos por otros distintos. Por ello se habla de metabolitos activos, o inactivos. Incluso, en ocasiones el fármaco no presenta actividad farmacológica alguna, siendo alguno de sus metabolitos los que realmente ejercen su actividad. Se habla en este caso de profármacos, y un ejemplo claro son algunas estatinas (simvastatina y lovastatina). Evidentemente, los profármacos dependen del buen funcionamiento del metabolismo para poder ejercer de forma adecuada sus efectos.
En ocasiones los propios fármacos o algunos de sus metabolitos son capaces de modificar la capacidad metabólica de las enzimas, aumentando o disminuyendo su actividad. Esta inducción o inhibición enzimática conlleva una mejoría o empeoramiento de la depuración de los fármacos, y subsecuentemente un posible aumento de su toxicidad o bien una disminución de su efecto. Este fenómeno es de gran trascendencia para algunas isoenzimas del citocromo p450, siendo objeto de continua investigación la determinación de los sustratos y de los inductores o inhibidores de las mismas.
La dotación enzimática viene determinada de forma genética, existiendo diferentes niveles de actividad en función del genotipo. Un ejemplo son los acetiladores lentos: sujetos que poseen una carga enzimática con menor capacidad para la metilación, por lo que en ellos son más frecuentes las interacciones y los casos de reacción adversa al fármaco. Estos son casi el 90% de la población japonesa, mientras que entre los europeos o los africanos están equilibrados con los acetiladores rápidos. Otros ejemplos pueden ser los metiladores rápidos, intermedios o lentos.
La farmacocinética estudia los mecanismos mediante los cuales se producen estas transformaciones, los tejidos en que ocurre, la velocidad de estos procesos y los efectos de las propias drogas y sus metabolitos sobre los mismos procesos enzimáticos. A modo de ejemplo, véase el diagrama del metabolismo hepático de los fármacos.
Excreción
Los fármacos son eliminados del organismo inalterados (moléculas de la fracción libre) o modificados como metabolitos a través de distintas vías. El riñón es el principal órgano excretor, aunque existen otros, como el hígado, la piel, los pulmones o estructuras glandulares, como las glándulas salivales y lagrimales. Estos órganos o estructuras utilizan vías determinadas para expulsar el fármaco del cuerpo, que reciben el nombre de vías de eliminación:- Orina,
- Lágrimas,
- Sudor
- Saliva
- Respiración
- Leche materna
- Heces
- Bilis
En otras ocasiones los fármacos son eliminados en la bilis con la que llegan hasta el intestino. Allí se unen a la fracción no absorbida del fármaco y se eliminan con las heces o bien pueden sufrir un nuevo proceso de absorción y ser eliminados finalmente por el riñón.
Las otras vías tienen poca transcendencia, salvo para fármacos muy concretos, como la vía respiratoria para el alcohol o los gases anestésicos, aunque en el caso de la leche materna es de especial trascendencia. El recién nacido presenta todavía cierta inmadurez de hígado o riñones y es más sensible a los efectos tóxicos del fármaco. Por ello hay que conocer qué fármacos pueden eliminarse a través de la leche materna para evitarlos.
Parámetros farmacocinéticos de la excreción
La farmacocinética estudia la forma y velocidad de depuración de los fármacos y sus metabolitos por los distintos órganos excretores, en relación con las concentraciones plasmáticas del fármaco. Para ello precisa de la definición operativa de algunos conceptos relativos a la excreción.Vida media
La vida media plasmática o vida media de eliminación es el tiempo necesario para eliminar el 50% del fármaco del organismo. O bien, el tiempo que tarda la concentración plasmática del fármaco en reducirse a la mitad de sus niveles máximos.Aclaramiento
Al medir la concentración plasmática de un fármaco antes de pasar por un órgano (sangre arterial) y después de haber pasado por él (sangre venosa) si se encuentra una diferencia de concentraciones se puede deducir que el órgano ha eliminado una parte del fármaco, aclarando la concentración del mismo. Desde esta óptica, se considera el aclaramiento como el volumen plasmático libre totalmente de fármaco por unidad de tiempo, por lo que se mide en unidades de volumen por unidades de tiempo. El aclaramiento, puede determinarse de una forma global («aclaramiento sistémico») o de forma individualizada para cada vía (aclaramiento hepático, renal, etc.). La ecuación que recoge este concepto sería: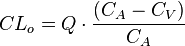
Es fácil comprender que cada órgano tendrá sus condicionantes del aclaramiento, en función de su mecanismo de acción para realizar la depuración. En lo que respecta al «aclaramiento renal», viene determinado por factores como el grado de unión a proteínas plasmáticas del fármaco, saturación de los transportadores (la secreción activa depende de proteínas transportadoras, que son saturables), o el número de nefronas funcionantes (de donde la importancia de situaciones como la insuficiencia renal).
En el caso del hígado, el «aclaramiento hepático» es fruto del metabolismo y por tanto está determinado por los factores que alteran el mismo así como por la cantidad de hepatocitos funcionantes, lo que justifica la importancia clínica de la insuficiencia hepática.
Estado de equilibrio
El estado de equilibrio o concentración estable es aquél en el que los aportes plasmáticos de fármaco se equilibran con la eliminación del mismo. Es fundamental su cálculo para decidir el período entre dosis y la cantidad de fármaco en cada una de ellas, en tratamientos prolongados.Otros parámetros de interés y ya vistos son la biodisponibilidad o el volumen aparente de distribución.
Farmacocinética Clínica
Gracias a la Farmacocinética Clínica, por ejemplo, se relanzó el empleo de ciclosporina como tratamiento inmunosupresor para posibilitar el trasplante de órganos sólidos (como el riñón), dado que tras demostrarse inicialmente sus propiedades terapéuticas se descartó prácticamente su uso por la nefrotoxicidad que provocaba en numerosos pacientes. Una vez que se comprobó que se podía individualizar la posología de la ciclosporina analizando las concentraciones plasmáticas de cada paciente (monitorización farmacocinética), la seguridad de este fármaco ha posibilitado gran cantidad de abordajes de trasplantes.
Clínicamente, la monitorización suele realizarse mediante la determinación de las concentraciones plasmáticas, ya que suele ser la determinación más accesible y fiable de las disponibles. Los principales criterios para determinar las concentraciones plasmáticas de un fármaco son:
- Estrecho intervalo terapéutico (intervalo entre las concentraciones tóxica y terapéutica)
- Alta toxicidad
- Elevado riesgo vital.
Medicamento
Un medicamento es uno o mas fármacos, integrados en una forma farmacéutica, presentado para expendio y uso industrial o clínico, y destinado para su utilización en las personas o en los animales, dotado de propiedades que permitan el mejor efecto farmacológico de sus componentes con el fin de prevenir, aliviar o mejorar enfermedades, o para modificar estados fisiológicos.Clasificación
Los medicamentos se dividen en:- farmacéutica: Es el medicamento de composición e información definidas, de forma farmacéutica y dosificación determinadas, preparado para su uso medicinal inmediato, dispuesto y acondicionado para su dispensación al público, con denominación, embalaje, envase y etiquetado uniformes según lo dispongan las autoridades sanitarias.
- Fórmula magistral: Es el medicamento destinado a un paciente individualizado, preparado por el farmacéutico, o bajo su dirección, para cumplimentar expresamente una prescripción facultativa detallada de las sustancias medicinales que incluye, según las normas técnicas y científicas del arte farmacéutico, dispensado en su farmacia o servicio farmacéutico y con la debida información al usuario.
- Preparado o fórmula oficinal: Es aquel medicamento elaborado y garantizado por un farmacéutico o bajo su dirección, dispensado en su oficina de farmacia o servicio farmacéutico, enumerado y descrito por el Formulario, destinado a la entrega directa a los enfermos a los que abastece dicha farmacia o servicio farmacéutico.
- Medicamento prefabricado: Es el medicamento que no se ajusta a la definición de especialidad farmacéutica y que se comercializa en una forma farmacéutica que puede utilizarse sin necesidad de tratamiento industrial y al que la autoridad farmacéutica otorgue autorización e inscriba en el registro correspondiente.
- Medicamento en Investigación: Forma farmacéutica de una sustancia activa o placebo, que se investiga o se utiliza como referencia en un ensayo clínico, incluidos los productos con autorización de comercialización cuando se utilicen o combinen, en la formulación o en el envase, de forma diferente a la autorizada, o cuando se utilicen para tratar una indicación no autorizada, o para obtener más información sobre un uso autorizado.
-
Según la prescripción médica
En España y algunos países latinoamericanos, los medicamentos se dispensan, distribuyen o venden exclusivamente en las farmacias. Existen dos tipos de medicamentos según la prescripción médica:
- Medicamento de venta libre: Son aquellos medicamentos que se distribuyen libremente en las farmacias, sin necesidad de receta o prescripción médica. Se dividen en dos categorías:
- Las Especialidades farmacológicas publicitarias (EFP) se corresponden con medicamentos publicitados en los medios de comunicación de masas como, por ejemplo, la televisión
- Los productos OTC ("Over the Counter") son fármacos destinados al alivio, tratamiento o prevención de afecciones menores, con los que se posee una amplia experiencia de uso y han sido expresamente autorizados como tales.
- Medicamento con receta médica: Son aquellos medicamentos recetados por un médico para el tratamiento de una enfermedad o síntoma en concreto.
-
Según derecho de explotación
- Medicamentos con patente: Aquellos medicamentos de investigación propia del laboratorio que los comercializa, sujetos a la protección comercial que brindan las agencias internacionales de patentes.
- Medicamentos genéricos: Aquellas presentaciones de moléculas que ya no están protegidas por la patente de su investigador.
Forma farmacéutica
Forma galénica o forma farmacéutica es la disposición individualizada a que se adaptan los fármacos (principios activos) y excipientes (materia farmacológicamente inactiva) para constituir un medicamento. O dicho de otra forma, la disposición externa que se da a las sustancias medicamentosas para facilitar su administración.El primer objetivo de las formas galénicas es normalizar la dosis de un medicamento, por ello también se las conoce como unidades posológicas. Al principio se elaboraron para poder establecer unidades que tuvieran una dosis fija de un fármaco con el que se pudiera tratar una determinada patología".
La importancia de la forma farmacéutica reside en que determina la eficacia del medicamento, ya sea liberando el principio activo de manera lenta, o en su lugar de mayor eficiencia en el tejido blanco, evitar daños al paciente por interacción química, solubilizar sustancias insolubles, mejorar sabores, mejorar aspecto, etc.
Historia
Es precisamente en la Edad Media donde comienza su actividad el farmacéutico separado del médico. En su botica realiza sus preparaciones magistrales, entendidas como la preparación individualizada para cada paciente de los remedios prescritos, y se agrupan en gremios junto a los médicos. En el renacimiento se va produciendo una separación más clara de la actividad farmacéutica frente a médicos , cirujanos y especieros, mientras que se va produciendo una revolución en el conocimiento farmacéutico que se consolida como ciencia en la edad moderna. La formulación magistral es la base de la actividad farmacéutica conjuntamente con la formulación oficinal, debido al nacimiento y proliferación de farmacopeas y formularios, y esta situación continua hasta la segunda mitad del siglo XIX.A partir de este momento empiezan a aparecer los específicos, que consistían en medicamentos preparados industrialmente por laboratorios farmacéuticos. Es así, que las formas galénicas no adquirirán verdadero protagonismo hasta alrededor de 1940, cuando la industria farmacéutica se desarrolla y éstas comienzan a fabricarse en grandes cantidades. Desde entonces hasta hoy en día las maneras en que se presentan los medicamentos han evolucionado y la diversidad que encontramos en el mercado es muy amplia.
Clasificación
Existen numerosas formas de clasificar las formas galénicas, según el factor que tengamos en cuenta: su estado físico, la vía de administración, el origen de sus componentes, etcétera. No obstante la más utilizada y la más útil desde el punto de vista de la medicina es la clasificación según la vía de administración que usen.De administración oral
La mayor parte de los fármacos administrados vía oral buscan una acción sistémica, tras un proceso previo de absorción entérica. En la absorción oral intervienen factores dependientes del individuo y otros dependientes de los fármacos que van a influir en la mayor o menor eficacia del fármaco administrado. Así mismo, la vía oral es motivo frecuente de interacciones farmacológicas, artículo éste que aconsejamos consultar para conocer la importancia de factores como el pH, toma o no de alimentos, tipo de éstos, velocidad del tránsito intestinal, u otros muchos que pueden influir en la absorción de un fármaco.La vía oral constituye la vía más utilizada de administración de fármacos, subdividiéndose a su vez, en formas líquidas y formas sólidas.
A.Formas orales líquidas. No plantean problemas de disgregación o de disolución en el tubo digestivo, lo que condiciona una acción terapéutica más rápida. Por el contrario no están protegidas, en caso de reactividad, frente a los jugos digestivos. Resultan de elección particularmente en niños. Los líquidos para administración oral son habitualmente soluciones, emulsiones o suspensiones que contienen uno o más principios activos disueltos en un vehículo apropiado. Los vehículos pueden ser:
- Acuosos: sirven para disolver principios activos hidrosolubles. Los más comunes son los jarabes (que contienen una alta concentración de azúcar, hasta un 64% en peso).
- Mucílagos: líquidos viscosos resultantes de la dispersión de sustancias gomosas (goma arábiga, tragacanto, agar, metilcelulosa) en agua. Se usan, sobre todo, para preparar suspensiones y emulsiones.
- Hidroalcohólicos: los elixires son soluciones hidroalcohólicas (25% alcohol) edulcoradas utilizadas para disolver sustancias solubles en agua y alcohol.
Las formas farmacéuticas líquidas para administración oral más usuales son:
Gotas
Son soluciones en las que el principio activo está concentrado.Jarabes
Forma farmacéutica que consiste en una solución acuosa con alta concentración de carbohidratos tales como sacarosa, sorbitol, dextrosa, etc.; de consistencia viscosa, en la que se encuentra disuelto el o los principios activos y aditivos.Tisanas
Las tisanas son infusiones con baja concentración de principios activos.
Elixires
Elixires de principios de siglo.
Suspensiones
Son mezclas heterogéneas formadas por un sólido en polvo (soluto) o pequeñas partículas no solubles (fase dispersa) que se dispersan en un medio líquido (dispersante o dispersora). Sólido en líquido que no es soluble en este.Suspensión extemporánea
Aquella que, por su poca estabilidad, se prepara en el momento de ser administrada.Viales bebibles
Comprimidos.
Comprimidos
Formas farmacéuticas sólidas que contienen, en cada unidad, uno o varios principios activos. Se obtienen aglomerando, por compresión, un volumen constante de partículas. Se administran generalmente por deglución, aunque se pueden dar otras posibilidades.Forma farmacéutica Cápsulas.
Cápsulas
Las cápsulas son preparaciones de consistencia sólida formadas por un receptáculo duro o blando, de forma y capacidad variable, que contienen una unidad posológica de medicamento. Este contenido puede ser de consistencia sólida, líquida o pastosa y estár constituido por uno o más principios activos, acompañados o nó de excipientes. El receptáculo se deshará por la acción de los jugos gástricos o entéricos, según la formulación, liberando entonces el principio activo.Granulados
Agregados de partículas de polvos que incluyen principios activos, azucares y coadyuvantes diversos. Se presentan en forma de pequeños granos de grosor uniforme, forma irregular y más o menos porosidad. Existen granulados de distintos tipos: efervescentes, recubiertos, gastrorresistentes y de liberación modificada.Sellos
Son cápsulas con un receptáculo de almidón. Prácticamente, han sido desplazados por las cápsulas duras.Píldoras
Preparaciones sólidas y esféricas, destinadas a ser deglutidas íntegramente. Cada unidad contiene uno o más principios activos interpuestos en una masa plástica. Se encuentran en franco desuso habiendo sido desplazadas por los comprimidos y cápsulas.Tabletas
Son pastillas para desleir en la cavidad bucal. Se diferencian de las píldoras por el tamaño y de los comprimidos por la técnica de elaboración. Sus constituyentes principales son la sacarosa, un aglutinante y uno o más principios activos.Pastillas oficinales o trociscos
Presentan una consistencia semisólida y están constituidas primordialmente por los principios activos y goma arábiga como aglutinante. Suelen recubrirse, para su mejor conservación, con parafina o azúcar en polvo (escarchado). Se emplean para la vehiculización de antitusígenos y antisépticos pulmonares.Liofilizados
Son preparaciones farmacéuticas que se acondicionan en forma de dosis unitarias y se liofilizan a continuación. Son formas muy porosas e hidrófilas y fácilmente dispersables en agua.Vía sublingual
Una forma especial de administración oral es la vía sublingual. En esta vía normalmente, se utilizan comprimidos que se disuelven debajo de la lengua absorbiéndose directamente. Tiene el inconveniente de ser exclusivamente permeable al paso de sustancias no iónicas, muy liposolubles. Esto hace que sólo puedan administrarse por esta vía fármacos de gran potencia terapéutica como la nitroglicerina o el isosorbide. Se utiliza para conseguir una acción terapéutica rápida o para fármacos que posean un alto grado de metabolización hepática, se degraden por el jugo gástrico o no sean absorbidos por vía oral. No obstante también se encuentran en el mercado presentaciones por comodidad del usuario. (Véase la formulación Flas en el epígrafe de innovaciones galénicas).De administración parenteral
La biodisponibilidad de un fármaco administrado vía parenteral depende de sus características fisicoquímicas, de la forma farmacéutica y de las características anatomofisiológicas de la zona de inyección:La vía intravenosa
Proporciona un efecto rápido del fármaco y una dosificación precisa, sin problemas de biodisponibilidad. Puede presentar, no obstante, graves inconvenientes, como la aparición de tromboflebitis, así como problemas de incompatibilidades entre dos principios activos administrados conjuntamente en la misma vía. Tiene el inconveniente de que no permite la administración de preparados oleosos debido a la posibilidad de originar una embolia grasa. Tampoco podrán usarse productos que contengan componentes capaces de precipitar algún componente sanguíneo o hemolizar los hematíes.La vía intraarterial
Utilizada en el tratamiento quimioterápico de determinados cánceres; permite obtener una máxima concentración del fármaco en la zona tumoral, con unos mínimos efectos sistémicos.La vía intramuscular
Se utiliza para fármacos no absorbibles por vía oral o ante la imposibilidad de administración del fármaco al paciente por otra vía ya que admite el ser utilizada para sustancias irritantes. Numerosos factores van a influir en la biodisponibilidad del fármaco por vía IM (vascularización de la zona de inyección, grado de ionización y liposolubilidad del fármaco, volumen de inyección, etc.). Esta vía es muy utilizada para la administración de preparados de absorción lenta y prolongada (preparados “depot”).La vía subcutánea
De características similares a la anterior pero al ser la piel una zona menos vascularizada, la velocidad de absorción es mucho menor. Sin embargo, dicha velocidad puede ser incrementada o disminuida por distintos medios. No puede utilizarse para sustancias irritantes ya que podría producir necrosis del tejido.Otras vías parenterales
De uso menos frecuente son la intradérmica, la intraaracnoidea o intratecal, epidural, intradural, intraósea, intraarticular, peritoneal o la intracardiaca.Los preparados para administración parenteral son formulaciones estériles destinadas a ser inyectadas o implantadas en el cuerpo humano. A continuación se enumeran cinco de las más representativas:
- Preparaciones inyectables. Son preparaciones del principio activo disuelto (solución), emulsionado (emulsión) o disperso (dispersión) en agua o en un líquido no acuoso apropiado.
- Preparaciones para diluir previamente a la administración parenteral. Soluciones concentradas y estériles destinadas a ser inyectadas o administradas por perfusión tras ser diluidas en un líquido apropiado antes de su administración.
- Preparaciones inyectables para perfusión. Son soluciones acuosas o emulsiones de fase externa acuosa, exentas de pirógenos, estériles y, en la medida de lo posible, isotónicas con respecto a la sangre.
- Polvo para preparaciones inyectables extemporáneas. Sustancias sólidas estériles, dosificadas y acondicionadas en recipientes definidos que, rápidamente tras agitación, en presencia de un volumen prescrito de líquido estéril apropiado, dan lugar a soluciones prácticamente límpidas, exentas de partículas, o bien a suspensiones uniformes.
- Implantes o pellet. Pequeños comprimidos estériles de forma y tamaño adecuados que garantizan la liberación del principio activo a lo largo de un tiempo prolongado.
De administración rectal
Supositorios
Supositorios y antíguos moldes de fabricación.
- Liposolubles: Son los más utilizados; entre ellos se encuentran la manteca de cacao, los glicéridos semisintéticos y los aceites polioxietilenados saturados.
- Hidrosolubles: polietilenglicoles (PEG).
Soluciones y dispersiones rectales
- Enemas: Son formas galénicas líquidas, de composición variable, destinadas a ser administradas por vía rectal, empleando para ello dispositivos especiales. Pueden tener como objetivo la vehiculización de un principio activo (enemas medicamentosos), el vaciado de la ampolla rectal (enemas evacuantes) o el administrar una sustancia radio-opaca para la realización de estudios radiológicos (enemas opacos).
- Colutorios.
Pomadas rectales
La vía rectal puede utilizarse para conseguir efectos locales o sistémicos. En este último caso, sólo se debe considerar como una alternativa a la vía oral cuando ésta no pueda utilizarse, ya que la absorción por el recto es irregular, incompleta y además muchos fármacos producen irritación de la mucosa rectal. Uno de los pocos ejemplos en los que esta forma farmacéutica tiene una indicación preferente es el tratamiento de las crisis convulsivas en niños pequeños (diacepam).También existen pomadas rectales para el tratamiento de las hemorroides externas.
De administración tópica
La vía tópica utiliza la piel y las mucosas para la administración de fármaco. Así pues, esto incluye mucosa ocular y genital. La mucosa oral ya ha sido vista dentro del epígrafe de la vía oral. La característica de esta vía es que se busca fundamentalmente el efecto a nivel local, no interesando la absorción de los principios activos. Los excipientes fundamentales para las formulaciones galénicas de uso tópico son tres: líquidos, polvos y grasas. Estos pueden combinarse entre sí de numerosas formas para adaptarse a las características del sitio en donde se van a aplicar, de ahí la variedad de formas galénicas para uso tópico.[7]| | | |
| | ||
| Pasta (Grasa+Polvo) | | Crema (Grasa+Líquido) |
| Tabla de tres entradas, con las posibles combinaciones entre Grasas, líquidos y polvos.[7] | ||
Baños
Consisten en la inmersión de todo o parte del cuerpo en una solución acuosa a la que se añaden determinados productos. Los más utilizados son los baños coloidales, que tanto tibios como calientes actúan como sedantes y antipruriginosos. En su preparación se mezcla una taza de almidón en un litro de agua y posteriormente esta preparación se hecha al agua del baño. En los baños oleosos se sustituye el almidón por aceites fácilmente dispersables, produciendo una suspensión homogénea. Actualmente se utilizan sobre todo los baños de sales, potenciados por el mundo de la cosmética.Lociones
También conocidas en dermatología como curas húmedas abiertas son muy útiles en inflamaciones superficiales como el eccema. Se colocan compresas empapadas en ciertos líquidos en las zonas secretantes y se cambian cada 5-10 minutos, o bien se añade de forma periódica líquido, de tal manera que se evite la desecación por efecto del calor de la piel. Esto se suele mantener durante 2 horas tres veces al día. El objetivo es el enfriamiento que consigue la evaporación del líquido, que actúa como antiinflamatorio. Además tienen propiedades antipruriginosas y vasoconstrictoras. Jamás irritan y no tienen riesgos en su uso. Los líquidos utilizados pueden ser el suero fisiológico, etanol al 10%, el permanganato potásico al 0.01% o la solución de Burow, compuesta por agua, subacetato de plomo y sulfato alumínico potásico. Una loción clásica de la farmacopea española sería el Agua de Dalibour, compuesta por una solución hidroalcohólica al 1% y sulfato de cobre, sulfato de cinc y alcanfor e indicada en el tratamiento del impétigo, dermatitis sobreinfectadas y dermatosis exudativas.
Toques o pincelaciones
Se suelen utilizar para descostrar heridas o lesiones dérmicas. Se utiliza el etanol al 10% al que se le puede añadir glicerina al 5%. El etanol puede causar quemazón, por lo que se desaconseja en región genital y cara. Una forma especial serían las pincelaciones secantes de la farmacopea alemana, que son en realidad lociones agitables, constituidas a partes iguales en peso de polvos y liquido, aunque son algo más cosméticas con una proporción algo inferior de polvos. A estas lociones se les puede añadir principos activos en forma de líquido o de polvo. Las indicaciones son las mismas que las de los polvos (ver después), ya que en realidad lo que se busca es el depósito de éstos sobre la piel al evaporarse el líquido de la formulación.Tinturas
Son preparaciones líquidas, coloreadas (de donde su nombre), resultado de la mezcla de hidroalcoholes con drogas secas a temperatura ambiente. Sus propiedades dependen del fármaco añadido, aunque en lineas generales son antisépticas y antipruriginosas. Lo más habitual suele ser el uso de soluciones de etanol, éter o cloroformo con productos como el mentol, fenol, fucsina, eosina, violeta de genciana o breas. Si resecan demasiado se puede añadir aceite de ricino al 1%. Se suelen administrar una vez al día y se retiran los restos al día siguiente antes de la nueva aplicación.Linimentos
Forma farmacéutica que consiste en una presentación líquida, solución o emulsión que contiene el o los principios activos y aditivos cuyo vehículo es acuoso, alcohólico u oleoso, y se aplica exteriormente en fricciones.Polvos
Los polvos se obtienen por división sucesiva de productos sólidos y secos hasta partículas homogéneas de tamaño variable. Tras su fabricación son pasados por tamices que oscilan entre los 0.1 mm de apertura del enrejado (polvos muy finos) hasta 1 mm. (polvos gruesos). Aplicados sobre la piel forman una capa finísima de propiedades refrescantes, vasoconstrictoras, antiinflamatorias y antipruriginosas. También protegen del roce entre superficies (pliegues) y ejercen protección mecánica. Los más famosos son los polvos de talco, que en dermatología se pueden asociar a otras sustancias como el óxido de zinc. También se cuenta con polvos de origen vegetal (almidón, licopodio), y en la industria cosmética se han utilizado ampliamente añadiendo pigmentos inócuos, del tipo del cinabrio, eosina, bentonita, óxido de hierro o ictiol para pieles oscuras. Aunque se pueden fabricar polvos directamente con las propias sustancias activas, no es esto lo más recomendable, siendo mejor su incorporación a los polvos inertes citados anteriormente. Así, se pueden añadir sustancias del tipo de las sulfamidas, fungicidas (ciclopirox olamina, miconazol), anhidróticos (cloruro de aluminio), etc. Como inconveniente importante tiene el que no se deben de utilizar ante presencia de secreciones, pues la mezcla con el exudado o el pus forma una especie de cemento que favorece la infección, es irritante y dificulta la cicatrización. Es especialmente desaconsejable la extendida utilización de polvos asociados a productos antipruriginosos en la varicela, donde pueden dejar cicatrices más intensas de lo habitual o la utilización de polvos de boro y ácido salicílico, muy extendidos por parte de la industria farmacéutica y de efectos no demostrados.Pastas
Están compuestas por polvos y grasas a partes iguales, aunque si son demasiado espesas se puede reducir la parte correspondiente a los polvos, aumentando la cantidad correspodiente de grasa. En líneas generales las pastas son secantes, pero mantienen la piel suave y plegable, protegiendo de traumas mecánicos y permitiendo la transpiración. Están indicadas en procesos secos (dermatitis crónicas) o muy poco secretantes y en la prevención de las úlceras de decúbito, por su efecto protector. Pueden incorporar principios activos tanto en forma líquida como sólida. Sus contraindicaciónes principales son las lesiones secretantes, infecciones y regiones pilosas. Se aplican extendiéndolas con una espátula de madera y se retiran con aceite de oliva o vaselina líquida. En lugar de grasas sólidas se pueden usar aceites, dando lugar a pastas oleosas. Si a una pasta se le añaden líquidos (agua), nos encontramos ante la pasta acuosa o pasta refrescante.Pomadas o ungüentos
Son grasas o sustancias de parecidas características que presenten aspecto semisólido a 25 °C. Es esta propiedad física lo que realmente las define ya que la composición química es enormemente variada. El término pomada es habitualmente usado en la literatura hispánica, mientras que el término ungüento es de uso preferente en el ámbito anglosajón, aunque por efecto de la preponderancia científica, éste se va extendiendo como forma principal de uso. Entre no entendidos se aplica el término pomada a cualquier presentación sólida de aplicación en la piel, sea crema, gel o pomada, de la misma forma que llaman pastilla a la mayoría de las formas galénicas de administración oral. Sus indicaciones son amplias: mejoran la piel seca y agrietada, son excelentes para retirar costras o escamas y son medianamente toleradas en zonas pilosas. Admiten multitud de principios activos bien en solución, los principios activos liposolubles, bien en forma de polvo o en dispersión coloidal para los insolubles en grasas. Su contraindicación más importante son los procesos inflamatorios agudos, tanto más contraindicada cuanto más húmedo sea el proceso.Geles
Son emulsiones semisólidas de polímeros orgánicos (metilcelulosa, agar, gelatina, propilenglicol, galato de propilo, edetato disódico, carboxipolímero) en un líquido (agua) Se suele añadir hidróxido sódico o ácido clorhídrico para ajustar el pH. Son, pues, bases incoloras, claras, no grasas, miscibles con agua. Se pueden considerar como un sistema coloidal donde la fase continua es sólida y la dispersa es líquida. En esta fase líquida puede incorporar principios activos en solución. A temperatura ambiente son sólidos o semisólidos fluidificándose al ser calentadas. Por su aspecto cosmético se suelen utilizar en zonas pilosas o estrechas (conducto auditivo externo, fosas nasales). Tras su aplicación desaparecen rápida y completamente.Colirios
Forma farmacéutica que consiste en una solución que contiene el o los principios activos y aditivos, aplicable únicamente a la conjuntiva ocular. Debe ser totalmente transparente, estéril, isotónica y con un pH neutro o cercano a la neutralidad.Gotas óticas y nasales
Las gotas óticas y nasales son una forma galénica para uso tópico consistente en un líquido acuoso u oleoso en el que van incorporados los principios activos y que se utilizan en el conducto auditivo externo o las fosas nasales respectivamente. Sus propiedades son similares a las de los colirios, salvo la exigencia de la esterilidad. Acorde a la gran cantidad de principios activos que tolera esta forma galénica, las indicaciones también son amplias:- Procesos infecciosos: Otitis externa, otitis de las piscinas, ectima nasal, etc.
- Procesos alérgicos, para el caso de las gotas nasales. La rinitis alérgica es una patología de alta prevalencia que en muchas ocasiones no es controlable con tratamiento sistémico.
- Procesos inflamatorios nasales (polipos nasales) u óticos.
- Patologías dermatológicas que afectan la piel del oido, como el psoriasis.
- Gotas nasales: Evitar sustancias que alteren la actividad de los cilios de las células de la mucosa nasal, como las sustancias boricadas.
- Gotas óticas: En lo posible evitar las soluciones oleosas y tener presente la posibilidad de que la membrana timpánica no esté indemne.
Apósitos
Cualquiera de los diferentes productos sanitarios empleados para cubrir y proteger una herida.Su finalidad última es la de promover la cicatrización de la herida. En función a ello, las características que ha de reunir un buen apósito son:- Impermeabilidad a los gérmenes, partículas y agua.
- Capacidad de absorción.
- Favorecimiento del pH ácido.
- Esterilidad.
- Permeabilidad a los gases.
- Proporcionar aislamiento térmico.
- Elasticidad y flexibilidad.
- Baja adherencia a la herida.
- Alto grado de cohesión.
- No tóxico.
- No alergizante.
Vía percutánea o transdérmica
A pesar de lo comentado respecto a la dificultad para la absorción de los principios activos, siempre existe la posibilidad de que se absorba una parte de los mismos a través de la piel. Este fenómeno se da con más intensidad en las zonas donde la piel está menos queratinizada (cara), donde se favorece la humedad (pliegues corporales) o en pieles más sensibles como la de los niños. Por otra parte, en ocasiones, sí que nos interesa que el principio activo sea absorbido para hacer su efecto a nivel sistémico. Esta vía, se conoce como vía percutánea o vía transdérmica y presenta unas características especiales.Parches transdérmicos
Forma galénica consistente en un reservorio con principio activo que se libera lentamente al aplicarlo sobre la piel. Se persigue que el fármaco pase a la circulación sistémica a través de la piel y no la actividad del fármaco en la propia piel. Estos parches proporcionan niveles plasmáticos terapéuticos constantes del fármaco, siempre que la piel permanezca intacta. Aunque relativamente recientes, tienen numerosas indicaciones y son objeto de continua investigación.La iontoforesis
Técnica utilizada en fisioterapia que consiste en la introducción de un fármaco o principio activo (mediante la utilización de corriente galvánica u otras formas de corrientes derivadas de esta) a través de la piel.Consiste en la colocación sobre la piel de dos electrodos que, por su polaridad, hacen que un fármaco cargado iónicamente atraviese la piel. Se produce por la interacción de la polaridad del electrodo con la carga iónica de la sustancia elegida (mediante el rechazo de los iones en el polo del mismo signo).
Las vías de introducción a través de la piel son sobre todo: folículos pilosos, glándulas sudoríparas y sebáceas, por interponer menor resistencia para el paso de sustancias.
De esta manera son administrados por vía percutánea fármacos cargados eléctricamente, sin producir efectos generales en el organismo, ya que sus efectos son locales.
De administración inhalada
Aerosoles: son dispositivos que contienen soluciones o suspensiones de un principio activo, envasadas en un sistema a presión de manera que, al accionar la válvula, se produce la liberación del principio activo en forma de microgotas impulsado gracias a un agente propelente. Los principios activos que pueden ser vehiculizados en esta forma galénica son numerosos:
-
- Agonistas β2 adrenérgicos:
- De Acción corta: salbutamol, levalbuterol, terbutalina y bitolterol.
- De acción prolongada: salmeterol, formoterol, bambuterol.
- Anticolinérgicos, como el bromuro de ipratropio.
- Glucocorticoides: beclometasona o fluticasona.
-
- Crisis asmática.
- Tratamiento de mantenimiento de la alergia respiratoria o asma extrínseca.
- En la enfermedad pulmonar obstructiva crónica (EPOC) tanto en el mantenimiento como en la profilaxis de las reagudizaciones.
- Sistema de nebulización por compresión.
- Inhaladores de polvo seco: a partir del medicamento en estado sólido, se liberan partículas suficientemente pequeñas de forma sincrónica con la inspiración; la fuerza de la inhalación arrastra el producto.
- Nebulizadores: son dispositivos que hacen pasar una corriente de aire sobre un líquido que lleva el principio activo en disolución. Se generan partículas uniformes y muy finas del principio activo (líquido) que son inspiradas junto con el aire que inyecta el nebulizador. Este sistema permite que el fármaco penetre más profundamente en las vías aéreas.
- Inhalaciones.
- Insuflaciones.
Innovaciones galénicas
La industria farmacéutica está continuamente desarrollando nuevas formas de hacer llegar el fármaco a su destino de la forma más rápida y eficaz. Al igual que el descubrimiento de nuevos fármacos, las nuevas galénicas están sujetas a patente de propiedad, pasando al cabo de un tiempo a dominio público. Así, por ejemplo, Norman Leo Henderson y Louis Nasir Elowe inventaron en 1969 las cápsulas de liberación prolongada con la patente 3427378 De esta manera, continuamente se están inventando sistemas, que basados en las fórmulas galénicas clásicas, aportan un plus de efectividad al producto comercial. Algunos ejemplos serían:- Sistema OROS o de Microbomba osmótica. Es un sistemas que permite la liberación temporal controlada del fármaco. Está constituido por un reservorio que contiene el fármaco, formado por un núcleo sólido con capacidad osmótica. Rodeando el reservorio existe una membrana semipermeable que permite el paso del agua procedente del exterior del sistema. Cuando el comprimido entra en contacto con el jugo gastrointestinal, la penetración del agua produce la disolución del núcleo osmótico y la salida del medicamento por un orificio o zona de liberación. El tamaño del poro de la membrana semipermeable va a condicionar la mayor o menor entrada de agua y, por tanto, la velocidad de liberación del principio activo.
- Sistema Filmtab.
- Sistema MUPS : En 1998 AstraZéneca desarrolló el sistema MUPS (Multiple Unit Pellet System) o Sistema Multigranular. La formulación de los comprimidos MUPS permite una liberación rápida de 1.000 a 2.000 unidades de principio activo con protección frente al ácido en el estómago. Son más pequeñas que las unidades contenidas en una cápsula tradicional, se dispersan con facilidad y se disuelven en el intestino delgadoofreciendo una eficacia mas predecible.
- Liposomas: Los liposomas son vesículas extraordinariamente pequeñas compuestas principalmente por fosfolípidos organizados en bicapas. Estas vesículas contienen una fase acuosa interna y están suspendidas en una fase acuosa externa. Se utilizan básicamente para transportar los principios activos de una manera lo más selectiva posible. Dependiendo de su naturaleza, el fármaco se puede incorporar dentro del liposoma (si es hidrofílico) o en la bicapa liposomal (caso de los lipofílicos).[26] Las ventajas de esta forma galénica serían:
- Aumento de la eficacia y disminución de la toxicidad del principio activo encapsulado.
- Prolongación del efecto.
- Mejor absorción, penetración y difusión.
- Posibilidad de vías de administración alternativas.
- Estabilización del principio activo.
- Sistema Flas: "Es una forma sólida oral que tiene como propiedad que no hace falta deglutir el contenido, sino que se absorbe directamente en contacto con la cavidad bucal"[2]
- Sistema Chronosphère.
- Gel Termoreversible: forma farmacéutica que permite que la preparación se mantenga en forma de solución líquida a temperaturas inferiores a 25 °C; en cambio a temperaturas próximas a 35 °C (cuando entra en contacto con el cuerpo humano) aumenta su viscosidad y la solución líquida se transforma en gel. Es ideal para uso en mucosas, ya que la forma líquida favorece la penetración pero al convertirse en sólido se evita el goteo y la deglución. Los laboratorios españoles SALVAT utilizan este sistema para algunos de sus productos de uso por vía nasal.
Producción
Los medicamentos son producidos generalmente por la industria farmacéutica. Los nuevos medicamentos pueden ser patentados, cuando la empresa farmacéutica ha sido la que ha investigado y lanzado al mercado el nuevo fármaco. Los derechos de producción o licencia de cada nuevo medicamento está limitado a un lapso que oscila entre 10 y 20 años. Los medicamentos que no están patentados se llaman medicamentos copia; en cambio, aquellos que no están patentados pero tienen un estudio de bioequivalencia, aprobado por las autoridades locales, se llaman medicamentos genéricos.Estudios de estabilidad
El objetivo de estos estudios es proveer evidencia documentada de como las características físicas, químicas, microbiológicas y biológica del medicamento varían con el tiempo bajo la influencia de factores como temperatura, humedad, luz y establecer las condiciones de almacenamiento adecuadas y el periodo de caducidad.La estabilidad de un fármaco se puede definie como la propiedad de éste envasasado en un determinado material para mantener durante el tiempo de almacenamiento y uso de las características físicas, químicas, fisicoquímicas, microbiológicas y biológicas entre los límites especificados.
Las formas de inestabilidad de un fármaco son:
- Química
- Física
- Biológica
Fechas de expiración
En el año 1979, en Estados Unidos se establecio una ley que pedia a las empresas productoras de medicamentos que pusieran la fecha hasta la cual se garantizaba la maxima potencia del medicamento, en otras palabras, no indica el momento en que yo es seguro tomarlas ni cuando se vuelven dañinas. La mayoria de medicinas son muy durables y se degradan lentamente, por lo que la mayoria, mientras haya sido apropiadamente fabricada, mantienen su potencia en altos rangos muy despues de su fecha de caducación. Un ejecutivo de Bayer, Chris Allen, dijo que las aspirinas que fabrican llevan una fecha de caducidad a los tres años luego de su fabricación, pero esta podria ser de incluso más y esta aun estaria en su maxima potencia, el motivo detras de esto yace en que constantemente estan mejorando la formula de las aspirinas, y hacer pruebas de caducidad de más de 4 años por cada actualización seria irrazonable. Sin embargo, se han registrado problemas con medicamentos que incluyen tetraciclina; tambien se hay excepciones en la duración, como son nitroglicerina, insulina, y algunos antibioticos líquidos.Reciclado
Una vez terminado el tratamiento para el que fueron prescritos, los medicamentos deben depositarse en un punto limpio, puesto que desecharlos indiscriminadamente junto con el resto de los residuos puede deteriorar gravemente el medio ambiente.Droga
En farmacología, una droga es toda materia prima de origen biológico que directa o indirectamente sirve para la elaboración de medicamentos, y se llama principio activo a la sustancia responsable de la actividad farmacológica de la droga. La droga puede ser todo vegetal o animal entero, órgano o parte del mismo, o producto obtenido de ellos por diversos métodos que poseen una composición química o sustancias químicas que proporcionan una acción farmacológica útil en terapéutica.Este término suele usarse inistintamente para designar a ésta y a los términos correspondientes en farmacia a principio activo, fármaco y medicamento, ya sea por extensión del concepto o debido a la traducción literal del término inglés drug, el cual no hace distinciones entre los tres conceptos.
Fármaco
Un fármaco, de acuerdo con la farmacología, es cualquier sustancia que produce efectos medibles o sensibles en los organismos vivos y que se absorbe, puede transformarse, almacenarse o eliminarse.
Esta definición se acota a aquellas sustancias de interés clínico, es decir aquellas usadas para la prevención, diagnóstico, tratamiento, mitigación y cura de enfermedades , y se prefiere el nombre de tóxico para aquellas sustancias no destinadas al uso clínico pero que pueden ser absorbidas accidental o intencionalmente; y droga para aquellas sustancias de uso social que se ocupan para modificar estados del ánimo.Los fármacos pueden ser sustancias creadas por el hombre o producidas por otros organismos y utilizadas por el hombre. De esta forma, hormonas, anticuerpos, interleucinas y vacunas son considerados fármacos al ser administrados en forma farmacéutica. En resumen, para que una sustancia biológicamente activa se clasifique como fármaco, debe administrarse al cuerpo de manera exógena y con fines médicos.
Los fármacos se expenden y utilizan principalmente en la forma de medicamentos, los cuales contienen el o los fármacos prescritos por un facultativo en conjunto con excipientes.
Patología
La patología es la parte de la medicina encargada del estudio de las enfermedades en su más amplio sentido, es decir, como procesos o estados anormales de causas conocidas o desconocidas.Las pruebas que mejor demuestran la existencia de una enfermedad se basan principalmente en el examen de una lesión en todos sus niveles estructurales, la evidencia de la presencia de un microorganismo (bacteria, parásito, hongo o virus) cuando se trata de una enfermedad infecciosa o la alteración de algún o algunos componentes del organismo (por ejemplo la glucosa en la diabetes mellitus, o la hemoglobina, en la anemia).
Los patólogos pueden ser anatomopatólogos o patólogos clínicos. Los anatomopatólogos se dedican al diagnóstico basado en la observación morfológica de lesiones, principalmente a través de la microscopía de luz, utilizando diversos tipos de tinciones. Los patólogos clínicos se dedican al diagnóstico a través de los análisis propios del laboratorio clínico, e incluye Hematología analítica, Inmunología diagnóstica, Microbiología diagnóstica, Bioquímica o Química clínica, Citogenética y Genética Molecular.
La patología no debe confundirse con la nosología, que es la descripción y sistematización de las enfermedades.
Muerte Celular
Cuando todos los mecanismos de adaptación y de resistencia se han agotado sobreviene la muerte celularLa célula puede morir de dos formas diferentes:
- Necrosis: comprende un estado irreversible de la célula, con incapacidad de mantenimiento de la integridad de la membrana plasmática y escapatoria de elementos citoplasmáticos, desnaturalización de las proteínas por autólisis o proveniente de enzimas líticas de leucocitos vecinos; ya que la necrosis atrae los componentes de la inflamación. Todos estos cambios condenan a la célula a perder su función específica, y solamente forma parte de restos celulares que serán fagocitados por los macrófagos.
- Apoptosis: Corresponde a cuando una célula, pierde el anclaje. Y por su uso, decide por así decirlo, "suicidarse". Reduciendo su citoplasma y fragmentando su material genético.
Apoptosis
La apoptosis es una forma de muerte celular, que está regulada genéticamente.La muerte celular programada es parte integral del desarrollo de los tejidos tanto de plantas (viridiplantae) como de animales pluricelulares (metazoa). En animales, la forma de muerte celular programada más corriente es la "apoptosis". Cuando una célula muere por apoptosis, empaqueta su contenido citoplasmático, lo que evita que se produzca la respuesta inflamatoria característica de la muerte accidental o necrosis. En lugar de hincharse o reventar y por lo tanto, derramar su contenido intracelular dañino enzimático, hacia el espacio extracelular-, las células en proceso de apoptosis y sus núcleos se encogen, y con frecuencia se fragmentan conformando vesículas pequeñas que contienen el material citoplasmático. De esta manera, pueden ser eficientemente englobadas vía fagocitosis y, consecuentemente, sus componentes son reutilizados por macrófagos o por células del tejido adyacente.
Inflamación aguda
La fase aguda de la inflamación es sinónimo de reacción inmune innata. En la inflamación aguda distinguimos tres puntos clave: cambios hemodinámicos, alteración de la permeabilidad vascular y modificaciones leucocitarias.Inflamación crónica
Cuando la inflamación se mantiene durante un tiempo prolongado (semanas o meses), se habla de inflamación crónica, en la que coexisten el daño tisular y los intentos de reparación, en diversas combinaciones, Puede producirse por mantenimiento de la inflamación aguda (si no se resuelve la causa), o bien empezar de manera progresiva y poco evidente, sin las manifestaciones de la inflamación aguda. Este segundo caso es el responsable del daño tisular de algunas de las enfermedades humanas más invalidantes, como la artritis reumatoide, la aterosclerosis, la tuberculosis o la fibrosis pulmonar. Además, es importante en el desarrollo del cáncer y en enfermedades que anteriormente se consideraban exclusivamente degenerativas, como el Alzheimer.En caso de no resolución se drenan también las bacterias y se extiende la infección por vía linfática: linfangitis (inflamación de los vasos linfáticos) y linfadenitis (inflamación de los ganglios linfáticos).
No hay comentarios:
Publicar un comentario